Skip to:
- Alzheimer’s Disease (AD)
- Parkinson’s Disease (PD)
- Huntington’s Disease (HD)
- Summary
Advances in stem cell technology have enabled us to study the mechanism of disease from cells taken directly from patients in vitro, called human induced pluripotent stem cells (hiPSCs).
Cellular models can provide deeper insights into the pathomechanisms of neurodegeneration that are difficult to investigate in vivo.
Other advances in cellular technology now enable researchers to edit the genome of cells using CRISPR-Cas9. This allows us to investigate how specific mutations (patient or de novo) affect the cells, and indeed in animal models. CRISPR-based genetic screens can also be achieved using iPSC derived neurons.
Alzheimer’s Disease (AD)
Alzheimer’s Disease (AD) is the most common form of dementia and is pathologically characterised by the presence of beta-amyloid plaques and neurofibrillary tangles of hyperphosphorylated tau. Together these contribute to the progressive neurodegeneration leading to cerebral and hippocampal atrophy, contributing to the progressive symptoms of dementia.
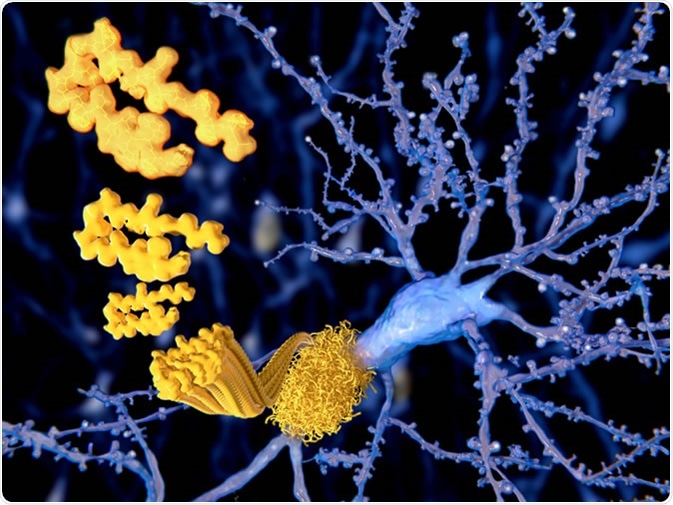
Most cases of AD are sporadic and have complex aetiologies. However, up to 10% of cases are due to genetically inherited mutations to genes that are involved in the production of beta-amyloid, including APP, PSEN1 and PSEN2. Aside from these genes, other risk genes have also been implicated in sporadic AD including APOE4, SORL1, and SMI-1. Other factors include the same risk factors for cardiovascular disease such as hypertension, atherosclerosis, and diabetes.
Patient-iPSC derived neurons have revealed many different mechanisms, depending on what the causative factor was in the patients. For example, PSEN1(P117R) mutations revealed neurons that were more susceptible to inflammation than controls. Furthermore, cells carrying mutations to PSEN1 were able to induce the hyperphosphorylation of tau. Other APP mutations including the V717 can also elevate levels of phosphorylated tau in neurons. The N1411 mutation in PSEN2 not only increased amyloid-beta (42) production, but also caused a decrease in action potentials. Treating patient-iPSC-neurons with β-/γ-secretase (PSEN1/2) inhibitors such as BSI-IV or compound E, has been shown to reduce beta-amyloid production, in particular the toxic 42-length form. Furthermore, treating cells derived from sporadic patients using β-secretase inhibitors βSi-II and OM99-2 also could reduce AD-phenotypes such as tau-phosphorylation, but not γ-secretase inhibitors.
Patient-iPSC derived neurons from sporadic AD cases with APOE4 alleles revealed high levels of phosphorylated-tau, higher levels of beta-amyloid production, and GABAergic neuronal degeneration. Thus, the mechanisms of sporadic AD are similar to familial AD. Furthermore, using an APOE4 structural corrector to mimic other protective forms of APOE, using PH002, AD phenotypes were reduced. These studies show not only the powerful application of using iPSCs to study disease mechanisms from different genetic variants, but also the ability to use those as drug screening factors in the assessment of various therapeutic compounds.
Parkinson’s Disease (PD)
PD is a common adult-onset neurodegenerative disease affecting 1% of the population over 60. At the centre of the pathology of PD, there is a loss of dopaminergic neurons in the substantia nigra, and is characterised by the presence of Lewy bodies containing aggregates of alpha-synuclein. The loss of these neurons leads to the motor symptoms of PD including tremor, rigidity, and postural instability.
About 10% of all PD cases are due to genetically inherited mutations involved in mitochondrial function and oxidative stress: SNCA, PARK2, PINK1, Parkin, DJ-1, and LRRK2. In most cases, PD is sporadic, though there are many risk factors including SNPs in SNCA, LRRK2, GBA1 and MAPT (tau). Environmental factors also play an important role in PD pathogenesis and include exposure to herbicides, pesticides, and heavy metals.
Most hiPSCs have focused on the genetic variants of PD, much like in AD. The cause of neuronal death has been attributed to mitochondrial dysfunction due to nitrosative and oxidative stress using SNCA(A53T) cells. Mechanistically, it has been shown that alpha-synuclein oligomers selectively induce oxidation of the ATP synthase beta-subunit leading to changes in permeability and subsequent cell death. In such mutations, mitochondria swell in dopaminergic and cortical neurons.
Another gene, LRRK2, is involved in both familial and sporadic PD. The G2091S mutation has been shown to upregulate alpha-synuclein and cause mitochondrial dysfunction in dopaminergic neurons derived from sporadic PD patients. Furthermore, the G2091S mutation of LRRK2 also has been shown to disintegrate the nuclear envelope, something characteristic of ageing. Blocking LRRK2 (by GW50764) has been shown to prevent neuronal death, and thus is a good candidate for drug therapy in PD.
Huntington’s Disease (HD)
Huntington’s Disease (HD) is a rare autosomal dominant progressive neurodegenerative disorder that has a single monogenic cause, a trinucleotide CAG polyglutamine repeat expansion within the huntingtin (HTT) gene. As a consequence, there is a toxic gain of function product that leads to death of GABAergic neurons within the striatum (striatal medium spiny neurons; MSNs). This leads to the motor symptoms of HD, including chorea and dystonia. CAG repeat length is correlated to disease severity, onset, and prognosis. Unlike most other neurodegenerative diseases that have numerous complex aetiologies, risk factors, and modifiers, HD is purely monogenic.
MSNs can be generated from patient derived hiPSCs. However, to induce a HD phenotype, cells often need to be exposed to stressors such as growth factor withdrawal, hydrogen peroxide treatment, or glutamate stimulation. These stressors can then lead to neuronal cell death and aggregation of mutant HTT.
Rodent studies have revealed that motor deficits can be rescued by transplanting human iPSC-derived neural stem cells, but not alter the function of the striatum due to the lack of effective MSN differentiation and the lack of striatal interneurons to support MSNs. Based on this and other observations that glial cells are important in HD pathogenesis, striatal transplantation of glia reduced disease pathology, and improving behavioural outcomes. Furthermore, glial transplantation also slowed disease progression and increased lifespan in rodent models. iPSC-based glial replacement may therefore be an important therapeutic strategy than iPSC-based MSN replacement in the treatment of HD.
Further support for glial-mediated therapy for HD was shown when a TNFa inhibitor, Xpro1595, was added to a co-culture of MSNs and astrocytes from HD-iPSCs. MSN death was reduced, as Xpro1595 reduced iNOS expression in astrocytes, therefore reducing glial mediated toxicity to MSNs. However, other iPSC studies also show that targeting pathogenic mechanisms in MSNs alone can also reduce HD phenotype, such as by activating DNA repair mechanisms (A2AR-PKA pathway to target H2AX phosphorylation) to improve cell viability.
Summary
Neurodegenerative diseases have many pathogenic overlaps in terms of oxidative stress, synaptic dysfunction, excitotoxicity, damaged DNA repair, mitochondrial dysfunction, toxic protein aggregates, and glial-support dysfunction. Patient-derived iPSCs from patients with neurodegeneration not only provide a tool for investigating intrinsic developmental and post-mitotic gene and protein expression, but also an excellent and simple drug screening mechanism. Furthermore, using CRISPR-Cas9 to edit faulty genes, or by using other therapeutic strategies outlined, cells can be transplanted back into the host as a stem cell based therapy, which has very promising results.
The development of 3D tissue cultures and organoids may improve our understanding of cell interactions in disease even further, and may provide more representative understanding of pathomechanisms of therapies that match the 3D environment of the brain. These provide an exciting and promising way to enhance iPSC cultures that can represent different regions of brain tissue. In these organoids, co-cultures of different cell types including neurons and glia can be made, and as long as adequate nutrients and oxygen is provided, 3D cultures can last much longer than 2D-cultures, and may be better for studying neurodegenerative processes that take time to form.
Whilst iPSC techniques have vastly improved and increased our knowledge and understanding of intricate cellular and molecular pathomechanisms of neurodegeneration, it is still important to progress such research; especially focusing on therapies, to animal models. Cell based therapy strategies in pre-clinical animal models are still needed to see if they can integrate, differentiation effectively and rescue both pathology and clinical outcomes. This is important as we first need to establish any ill-effects of iPSC transplantation before trialling them in human patients.
Sources
- Wu et al, 2019. Opportunities and challenges for the use of induced pluripotent stem cells in modelling neurodegenerative disease. Open Biol. 9(1): 180177. https://www.ncbi.nlm.nih.gov/pubmed/30958120
- Kampmann, 2017. A CRISPR Approach to Neurodegenerative Diseases. Trends Mol Med. 23(6):483-485. https://www.ncbi.nlm.nih.gov/pubmed/28478951
Further Reading
- All Stem Cells Content
- Stem Cell Sources, Types, and Uses in Research
- What are Stem Cell Transplants?
- Stem Cell Properties
- What are Stromal Cells?
Last Updated: Oct 27, 2019

Written by
Osman Shabir
Osman is a Neuroscience PhD Research Student at the University of Sheffield studying the impact of cardiovascular disease and Alzheimer's disease on neurovascular coupling using pre-clinical models and neuroimaging techniques.
Source: Read Full Article